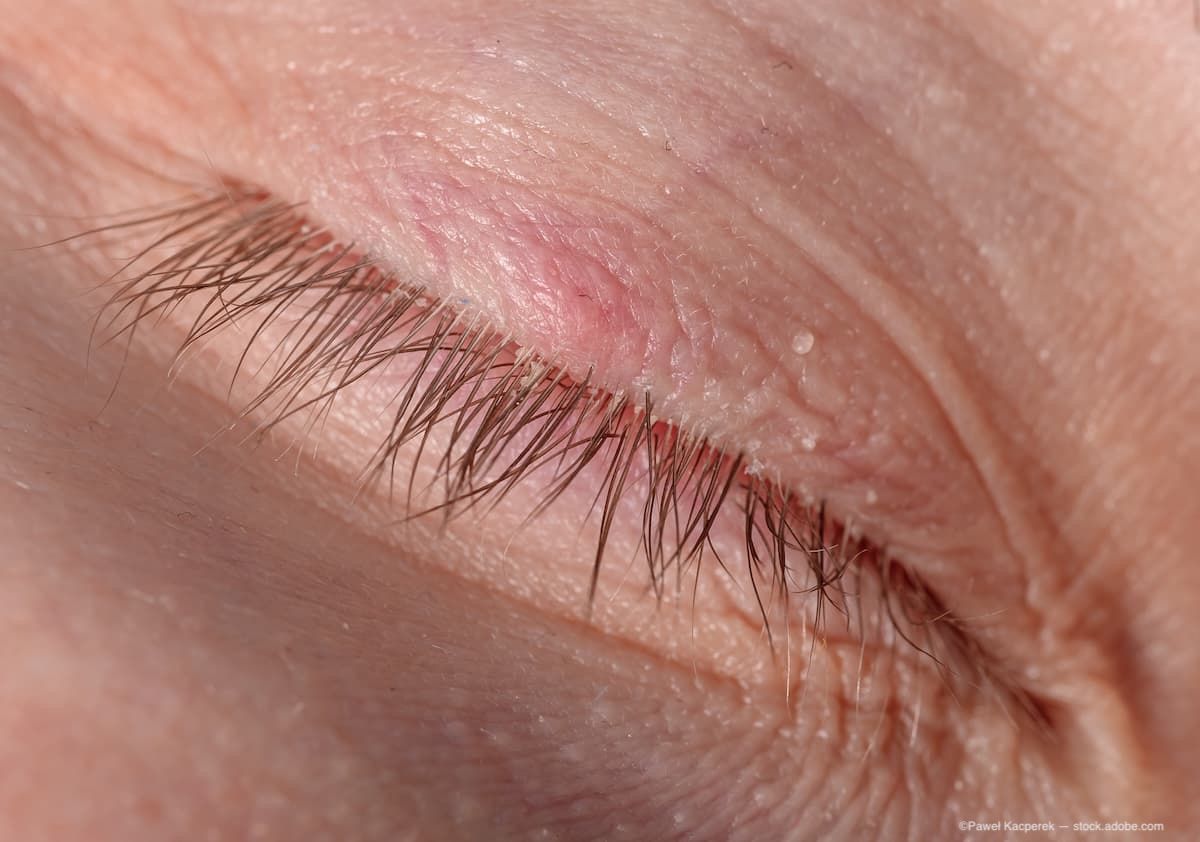
Управление по контролю качества пищевых продуктов и медикаментов США (FDA) приняло повторную заявку компании Aldeyra Therapeutics на регистрацию лекарственного препарата репроксалап для лечения сухого глаза (DEA). Установленная дата рассмотрения заявки — 16 декабря 2025 года.
Повторная подача заявки последовала после получения компанией второго уведомления FDA о полном отказе (Complete Response Letter, CRL), отправленного три месяца назад. В уведомлении указывались методологические проблемы в ранее проведенном клиническом исследовании с использованием сухого глазного аппарата, включая дисбаланс базовых показателей среди групп лечения, который мог повлиять на интерпретацию результатов эффективности терапии.
Компания Aldeyra представила обновленную заявку, включив данные нового клинического исследования, устраняющие выявленные FDA недостатки и подтвердившие достижение основной точки оценки эффективности репроксалапа.
В мае 2025 года компания сообщила о положительных результатах фазы III двойного слепого, рандомизированного контролируемого испытания, проведенного с применением репроксалапа. Исследование показало статистически значимое снижение дискомфорта глаз участников опытной группы (n = 58) по сравнению с контрольной группой (n = 58) через 80–100 минут после начала эксперимента. Разница средних квадратичных отклонений составила 6,5 (доверительный интервал 95%: −10,5 до −2,5; р-значение = 0,002), что подтверждает эффективность препарата в моделировании острого приступа сухого глаза. Не было выявлено серьезных побочных эффектов, наиболее распространенной нежелательной реакцией была легкая кратковременная боль в месте введения препарата, которая исчезала менее чем за одну минуту.
Президент и генеральный директор Aldeyra доктор Тодд К. Брейди отметил, что, следуя рекомендациям FDA относительно проведения дополнительного клинического исследования, новая заявка содержит единственное исследование, которое успешно достигло поставленной цели снижения дискомфорта глаз по сравнению с контролем.